

Defective Drugs
FDA Reports Top Consumer Updates of 2012
 Disposal of unused drugs, dangerous diet supplements and contaminated tattoo ink led the health and safety topics which most interested consumers in 2012.
Disposal of unused drugs, dangerous diet supplements and contaminated tattoo ink led the health and safety topics which most interested consumers in 2012.
The Food and Drug Administration (FDA) reported these were among the top 10 most read topics on its Consumer Updates, which can be found on its website and are distributed through e-mail subscriptions.
How to Dispose of Unused Medicines. Consumers should follow any directions on the label for proper disposal. Otherwise, most prescription drugs can be thrown in the trash, unless the directions specify they should be flushed down the toilet. Drugs thrown in the trash should be mixed with substances such as coffee grinds, kitty litter or leftover food. Consumers should also call their city or municipal office and ask about community drug take-back programs that allow the public to bring unused drugs to a central location for safe disposal. Read more.
Mercury Poisoning Linked to Skin Products. Federal health officials warn consumers to beware and avoid use of skin products containing mercury. No product brand name was released, but consumers should check the product ingredients for “mercurous chloride,” “calomel,” “mercuric,” “mercurio,” or “mercury,” and stop using the product immediately. These products are manufactured in other countries and have been illegally sold in the United States. They have been found in at least seven states. Mercury poisoning can cause a wide range of problems, including memory problems, depression, tremors and changes in vision or hearing. You can suffer poisoning through use of these creams, as well as by breathing the vapors if you live with someone who uses one. Read more.
HCG Diet Products Are Illegal. The FDA warns consumers to avoid unsafe “homeopathic” human chorionic gonadotropin (HCG) weight-loss products. HCG is approved by FDA as a prescription drug for the treatment of female infertility and other medical conditions, but is not approved for weight loss or over the counter treatment for any use. Read more.
Warning on Hydroxycut Products. Consumers are urged not to use Hydroxycut products by lovate Health Sciences Inc., of Oakville, Ontario. The company agreed to recall the dietary supplements which were associated with a number of serious liver injuries. They are marketed for weight loss, as fat burners, as energy-enhancers, as low carb diet aids, and for water loss under the Iovate and MuscleTech brand names. Read more.
Examining Arsenic in Rice: A Work in Progress. The FDA is studying the level of arsenics in rice and rice products, which includes rice as well as foods rice is processed into such as cereals, rice beverages and rice cakes. Arsenic levels have been found in various rice products, but the FDA is not recommending consumers stop consumption at this time. It is advising consumers eat a balanced diet containing a wide variety of grains. Read more.
Tattoo Inks Pose Health Risks. The FDA is investigating after contaminated tattoo inks have caused serious infections in at least four states in 2011 and 2012, including in 19 people in New York and others in Iowa, Washington and Colorado. The agency is particularly concerned about a family of bacteria called nontuberculous Mycobacteria (NTM) that has been found in a recent outbreak of illnesses linked to contaminated tattoo inks. M. chelonae, one of several disease-causing NTM species, can cause lung disease, joint infection, eye problems and other organ infections. Read more.
Triclosan: What Consumers Should Know. The FDA has engaged in a comprehensive scientific and regulatory review of triclosan, which has been found in many antibacterial soaps, body washes and toothpastes. The agency is not advising consumers to avoid use at this time, but states those who are concerned should check labels to see if it is contained in products they are using. Read more.
FDA Expands Caution About SimplyThick. The FDA advises that infants may face an increased risk of developing a life-threatening condition if they are fed a thickening product called Simply Thick. The agency has identified 22 infants who have developed necrotizing enterocolitis (NEC) since May 2011, seven of whom have died. It says further study is needed to determine if there is an actual link between the thickening product and the condition. Read more.
FDA Expands Advice on Statin Risks. Consumers who take statins to prevent heart disease are given new recommendations, including that cognitive impairment such as memory loss has been reported by some statin users. The FDA also says routine monitoring of liver enzymes in the blood has been found to be ineffective and is no longer needed. Read more.
Treating Cushing’s Disease in Dogs. An overview of how veterinarians commonly treat the illness and what dog owners should know. Read more.
The Boston personal injury lawyers at Breakstone, White & Gluck are experienced in handling cases involving product defects and medical device recalls. If you have been injured, it is important to learn your rights. Contact our injury lawyers toll-free at 800-379-1244 or 617-723-7676 or use our contact form.
Meningitis Outbreak: Over 300 Now Infected, 23 Dead as State Calls for Licenses
State officials have announced they will seek permanent surrender of New England Compounding Center’s (NECC) pharmacy license in the wake of a fungal meningitis outbreak which has infected more than 300 people and left 23 dead.
At a Tuesday press conference, state officials announced they will seek the pharmacy’s license and permanent license revocation for NECC’s three pharmacists. Recent state inspections of the closed-down Framingham facility found dirty and defective equipment and showed the company had failed to follow its own safety tests and properly sterilize tools.
Governor Deval Patrick said a federal criminal investigation has been launched, while Madeleine Biondolillo, director of the state Bureau of Health Care Safety and Quality, said the company had been violating its state compounding license. The facility was licensed to produce medicine with a patient-specific prescription in hand, not manufacture large shipments.
As of this week, 304 people had contracted fungal meningitis or joint infections linked to the injectable steroid produced by NECC, reported The Boston Globe. Testing has found the fungus matches a type found in unopened vials of the steroid shipped from NECC, though they are still investigating how the fungus got into the vials. The injectable steroid is methylprednisolone acetate.
The 23 deaths linked to the defective steroid come from seven states. No one has been reported ill in Massachusetts. But federal officials say as many as 14,000 people may have been exposed to the contaminated drug between May 21 and Sept. 26, when it was recalled. The company went onto recall all its drugs.
The Food and Drug Administration released a list this week of more than 3,000 NECC customers who received products other than the injectable steroid blamed for the meningitis outbreak. They included 215 Massachusetts doctors, health centers and hospitals. The shipments fell within the time frame of when the defective steroids were produced at the facility.
Ameridose, a Westborough pharmacy with the same owners as NECC, has also been closed down since Oct. 10 in an agreement with state officials. It is scheduled to remain closed until Nov. 5 for state inspections.
Related:
- CDC: 10 more people have fungal infections linked to Framingham pharmacy, The Boston Globe.
- New England Compounding did not follow sterility procedures, investigators find; state orders regular inspections of similar pharmacies, The Boston Globe.
- Corrected list of New England Compounding customers includes 215 in Massachusetts, The Boston Globe.
Contaminated Steroid Injections Trigger Outbreak in 23 States, 5 Deaths
Five people have died and at least 35 have fallen ill after being injected with a steroid which was contaminated with fungus from a Massachusetts compounding pharmacy.
The steroid was shipped to 75 health centers in 23 states, not including Massachusetts, according to media reports. Patients treated with the steroid in six states have fallen ill and died from aspergillus meningitis. Symptoms of this rare infection include headache fever, nausea and pain at the injection site.
The steroid was produced at New England Compounding Center of Framingham, Massachusetts. The company first recalled three lots of methylprednisolone acetate last week. This week, the Food and Drug Administration (FDA) has expanded the recall to include all injectable spinal drugs made by New England Compounding Center. The FDA is further urging health care providers to discard all products from the company as a precaution.
The 35 people who were stricken ill were treated with epidural injections of methylprednisolone acetate for lower back pain between July and September. The infected patients are from Tennessee, Virginia, Maryland, Florida, North Carolina and Indiana. Patients are still being notified. No Massachusetts facilities received the defective steroid injections, but there were shipments within New England to health care providers in Connecticut, Rhode Island and New Hampshire.
Nine Massachusetts health facilities did receive methylprednisolone acetate from other lots made by New England Compounding Center, but no cases of fungus meningitis have been reported.
New England Compounding Center has voluntarily given up its state license and ceased operation. Investigators this week found a contaminated sealed vial of steroid at the company, the FDA reported, and testing is being performed to determine if it is the same fungus as the outbreak.
The Boston Globe reported a state official said the Massachusetts Board of Registration in Pharmacy received complaints about the company in 2002 and 2003. The board worked with the company to improve conditions, but the official did not share details. The board is still investigating another complaint received in March about eye medications.
In 2006, the FDA sent the company a letter stating concerns including its splitting and repacking of the injectable colorectal cancer drug Avastin.
The case shines a light on regulations regarding pharmacies which prepare custom medications, often for patients who have allergies to other medications or for treatments which are no longer commercially available.
New England Compounding Center was not accredited by the Pharmacy Compounding Accreditation Board, which conducts a site visit and review of pharmacy every three years. The accreditation is voluntary.
Related:
5 deaths now tied to rare fungal meningitis possibly contracted from steroids prepared by Massachusetts pharmacy, Boston Globe.
Hundreds seen at risk in meningitis outbreak, Associated Press.
Massachusetts Board of Registration in Pharmacy.
Defective Drug Warning: Reumofan Diet Supplements
 Those who take so-called “natural dietary supplements” want to listen to the Food and Drug Administration’s (FDA) latest alert on Reumofan Plus and Reumofan Plus Premium, which have now been linked to cases of death and stroke.
Those who take so-called “natural dietary supplements” want to listen to the Food and Drug Administration’s (FDA) latest alert on Reumofan Plus and Reumofan Plus Premium, which have now been linked to cases of death and stroke.
“Consumers who are currently taking Reumofan Plus or Reumofan Plus Premium, or who have recently stopped taking it, should immediately consult a health care professional,” said Elizabeth Miller, Pharm.D., acting director of FDA’s Division of Non-Prescription Drugs and Health Fraud.
The FDA issued its first alert on the dangers of Reumofan Plus on June 1, stating the drug had several active pharmaceutical ingredients not listed on the label. This week, the agency issued an updated alert for both the drug and Reumofan Plus Premium, saying it had since received reports of death, stroke and these symptoms:
- Severe bleeding in the gastrointestinal tract
- Dizziness
- Insomnia
- High blood sugar levels
- Problems with liver and kidney functions
- Corticosteroid withdrawal syndrome
These defective products are marketed as natural dietary supplements which treat arthritis, muscle pain, osteoporosis and bone cancer. If not properly supervised by a medical professional, those who stop taking the supplements can face serious withdrawal symptoms.
The defective drugs may contain corticosteroids and abrupt discontinuation can supress the adrenal glands, which regulate a number of hormone and body functions. Users could experience fatigue, low blood sugar levels, fainting and other symptoms.
The Reumofan products are manufactured in Mexico by Riger Naturals and sold on the Internet, flea markets and some retail outlets. The Mexican Ministry of Health has also issued a health warning to its citizens and ordered Riger Naturals to recall the products.
FDA testing of Reumofan Plus and Reumofan Plus Premium show they contain two ingredients which require prescriptions: diclofenac sodium and methocarbamol. Reumofan Plus also contained dexamethason, a type of corticosteroid.
The FDA offers consumers these tips for avoiding defective and dangerous drugs:
- Be wary of off-the-counter products which only have prescription counterparts.
- Be cautious when a label has a foreign language.
- Be aware that so-called “natural dietary supplements” are not subject to the same FDA pre-market review as prescription drugs and some over-the-counter drugs.
Related:
- Reumofan Products Pose Risk to Consumers, Food and Drug Administration.
- Dietary Supplements, Food and Drug Administration.
Recalled Birth Control Pills: Lo/Ovral-28, Norgestrel and Ethinyl Estradiol
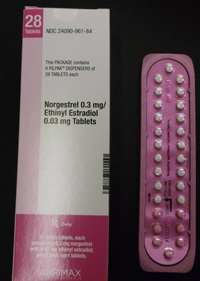 Pfizer Inc. has voluntarily recalled certain lots of birth control pills which may contain ingredient errors or out-of-sequence packaging which could have exposed women to a risk for unintended pregnancy.
Pfizer Inc. has voluntarily recalled certain lots of birth control pills which may contain ingredient errors or out-of-sequence packaging which could have exposed women to a risk for unintended pregnancy.
In January, Pfizer recalled 14 lots of Lo/Ovral-28 (norgestrel and ethinyl estradiol) Tablets and 14 lots of Norgestrel and Ethinyl Estradiol Tablets (generic) for customers in the U.S. market. The defective pills were distributed to warehouses, clinics and retail pharmacies nationwide.
An investigation by Pfizer found that some blister packs may contain an inexact count of inert or active ingredient tablets and that the tablets may be out of sequence. Pfizer recalled the tablets on January 31, 2012, with knowledge of the Food and Drug Administration (FDA). Pfizer said the error has been corrected.
The tablets were manufactured and packaged by Pfizer Inc., commercialized by Akrimax Rx Products and labeled under the Akrimax Pharmaceuticals brand. The medicine is packaged in blister packs of 21 tablets of active ingredients and seven tablets of inert ingredients. Click the link below for packaging numbers involved in the recall.
The product liability lawyers at Breakstone, White & Gluck are reviewing cases for women who have taken defective lots of these birth control pills and have experienced or are experiencing an unplanned pregnancy. Contact us today at 800-3791244 or 617-723-7676 or use our contact form. Read More
Defective Medication Under Scrutiny After Supreme Court Ruling
A recent Supreme Court ruling is limiting court actions by injured patients who have filed claims against manufacturers of generic drugs.
The ruling was issued last year and said generic drugmakers do not have control over their labels and therefore cannot be sued for failing to alert the public. Under the 1984 Hatch-Waxman Act, generic drugmakers were not required to undergo the Food and Drug Administration’s (FDA) lengthy approval process if they could prove the generic drug was equivalent to the brand-name medicine.
In most cases, the Henry-Waxman Act requires generic manufacturers use the same labels as brand-name drugs, with dosing instructions and risks for injury. For this reason, judges have started to dismiss many product liability lawsuits against generic manufacturers while allowing those against brand-name drugs to move ahead.
In a March 20, 2012 article, The New York Times reported that a woman who had received the brand name for an anti-nausea medication had suffered gangrene – or a condition that results in dead or weakening body tissue. She sued the manufacturer Wyeth and won $6.8 million.
Another woman took the generic version of the defective drug, known as promethazine, and had to have her arm and forearm amputated because of complications from gangrene. Her case was dismissed last fall following the Supreme Court ruling.
The Supreme Court ruling comes as Americans are increasingly turning to generic medicines. As prices skyrocket and the economy struggles, many health insurance companies are requiring generics be filled before brand-name drugs. Doctors are required to show medical needs for the brand name over generic.
As a result, nearly 80 percent of prescriptions in the United States are filled generic and most states permit pharmacists to dispense a generic in place of a brand name.
What Can Consumers Do:
Support efforts to change the law. Public Citizen, a consumer advocacy group, has petitioned the FDA to give generic companies greater control over their labels. The move may allow generic drug users to sue. U.S. Rep. Henry A. Waxman, D-California, is also exploring ways to address the issue.
Talk to your doctor. Ask your doctor about the medicine being prescribed, the generic and potential side effects. If you are still concerned about potential injuries, ask your doctor to call your insurance company and request a brand-name.
Research any medication you use. Write down the name of the medicine you are prescribed, the medicine you receive at the pharmacy and research both drugs. Discuss any side effects with your physician.
Consider foregoing insurance. If you are really concerned and can afford the brand-name prescription, consider purchasing it. There are many discount drug programs which may help you reduce your costs. Check with any groups you are affiliated with, including AAA and AARP.
Contact your health insurance company. If the company has required you to use generic medications, ask if it has changed its policy and is now allowing use of brand-name medications.
Related:
Massachusetts Patients’ Bill of Rights and Preventing Medical Errors.
Multaq Heart Drug Warning from FDA
 After years of close monitoring and label changes for Multaq, the Food and Drug Administration (FDA) has issued a new warning that the heart drug places some patients at an increased risk for serious cardiac events including death.
After years of close monitoring and label changes for Multaq, the Food and Drug Administration (FDA) has issued a new warning that the heart drug places some patients at an increased risk for serious cardiac events including death.
Multaq is used to treat various conditions, including patients with permanent Atrial Fibrillation (permanent AF). This is the most serious form of AF, which is an abnormal heart rhythm. The two other types are paroxysmal AF and persistent AF.
Permanent AF is a chronic condition in which sinus rhythm cannot be sustained despite treatment.
The FDA issued its Dec. 19, 2011 safety notification based on its monitoring of a 10-year study. In the communication, the FDA stated Multaq doubles the rate of cardiovascular death, stroke and heart failure in patients with permanent AF. The FDA advised healthcare professionals to:
- Not prescribe Mutlaq to patients with AF who cannot or will not be converted into normal sinus rhythm (permanent AF).
- Monitor heart rhythm by electrocardiogram at least once every three months.
- If the patient is in permanent AF, Multaq should be stopped or the patient should be cardioverted.
- Multaq is indicated to reduce hospitalization for AF in patients in sinus rhythm with a history of non-permanent AF.
- Patients prescribed Multaq should receive antithrombatic therapy.
The study the FDA acted on was called called the Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS). The study was stopped early in July 2011. The FDA said it will release more information as it becomes available.
Multaq Background
Dronedarone is the generic name for Multaq. It was approved by the FDA in 2009 to treat several conditions, including permanent AF. Manufactured by Sanof-Aventis, the drug has been prescribed to 500,000 people around the world.
Since 2009, Multaq has been subject to several FDA actions, including in 2010 a revised warning noting cases of worsening heart failure in some patients. In February 2011, the FDA changed the warning label to state that Multaq should be discontinued if liver damage is suspected.
Related Blogs and Websites
- Actos: FDA Warning About Bladder Cancer
- DePuy Hip Recall: Insurers Seek to Collect Settlements
- Transvaginal Mesh: What to Know If You’ve Been Injured
Actos Warning From FDA
Hundreds of thousands of diabetes patients have been re-considering their medication plans after a recent warning from the Food and Drug Administration (FDA).
On June 15, 2011, the FDA issued a safety communication reporting that use of the oral diabetes medication Actos (pioglitazone) for more than one year may be associated with an increased risk of bladder cancer.
The FDA warned that patients with active bladder cancer should not be taking pioglitazone and those with a prior history should only use it after considering the potential medical benefits against the risk for cancer recurrence.
Actos is an oral drug used to treat type II diabetes, the most common form of the illness that plagues 25.8 million Americans. Actos is manufactured by Takeda Pharmaceutical Company and co-marketed by Eli Lilly and Company. It is one of the most widely used medications on the market. Over 2 million patients filled prescriptions for it in 2010.
The communication came as the FDA placed restrictions on Avandia, another popular medication, after studies showed it increased the risk of heart attacks. Both drugs belong to the thiazolidinedione class of drugs.
The warning about Actos came as the FDA made a periodic review of an ongoing 10-year Actos study into the risk for developing bladder cancer. The study reports there may be a 40 percent greater risk for developing bladder cancer among those who take Actos for more than a year.
The FDA’s warning came a new epidemiological study out of France suggested an increased risk of bladder cancer with pioglitazone use. As a result, France has suspended use of Actos and Germany has recommended new patients not be placed on the drug.
On August 4, 2011, the FDA reported it had approved new label and medication instructions for Actos. The FDA is continuing to monitor the results of Actos cancer studies.
Patients taking Actos should contact their doctors if they experience any sign of blood in the urine or a red color in the urine or other urinary pain, as these Actos complications may be signs of bladder cancer.
Click here to read the June 15, 2011 FDA Safety Communication on Actos.
Read More