

Products Liability
Recalled Birth Control Pills: Lo/Ovral-28, Norgestrel and Ethinyl Estradiol
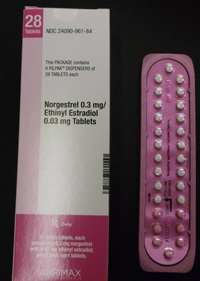 Pfizer Inc. has voluntarily recalled certain lots of birth control pills which may contain ingredient errors or out-of-sequence packaging which could have exposed women to a risk for unintended pregnancy.
Pfizer Inc. has voluntarily recalled certain lots of birth control pills which may contain ingredient errors or out-of-sequence packaging which could have exposed women to a risk for unintended pregnancy.
In January, Pfizer recalled 14 lots of Lo/Ovral-28 (norgestrel and ethinyl estradiol) Tablets and 14 lots of Norgestrel and Ethinyl Estradiol Tablets (generic) for customers in the U.S. market. The defective pills were distributed to warehouses, clinics and retail pharmacies nationwide.
An investigation by Pfizer found that some blister packs may contain an inexact count of inert or active ingredient tablets and that the tablets may be out of sequence. Pfizer recalled the tablets on January 31, 2012, with knowledge of the Food and Drug Administration (FDA). Pfizer said the error has been corrected.
The tablets were manufactured and packaged by Pfizer Inc., commercialized by Akrimax Rx Products and labeled under the Akrimax Pharmaceuticals brand. The medicine is packaged in blister packs of 21 tablets of active ingredients and seven tablets of inert ingredients. Click the link below for packaging numbers involved in the recall.
The product liability lawyers at Breakstone, White & Gluck are reviewing cases for women who have taken defective lots of these birth control pills and have experienced or are experiencing an unplanned pregnancy. Contact us today at 800-3791244 or 617-723-7676 or use our contact form. Read More
Defective Medication Under Scrutiny After Supreme Court Ruling
A recent Supreme Court ruling is limiting court actions by injured patients who have filed claims against manufacturers of generic drugs.
The ruling was issued last year and said generic drugmakers do not have control over their labels and therefore cannot be sued for failing to alert the public. Under the 1984 Hatch-Waxman Act, generic drugmakers were not required to undergo the Food and Drug Administration’s (FDA) lengthy approval process if they could prove the generic drug was equivalent to the brand-name medicine.
In most cases, the Henry-Waxman Act requires generic manufacturers use the same labels as brand-name drugs, with dosing instructions and risks for injury. For this reason, judges have started to dismiss many product liability lawsuits against generic manufacturers while allowing those against brand-name drugs to move ahead.
In a March 20, 2012 article, The New York Times reported that a woman who had received the brand name for an anti-nausea medication had suffered gangrene – or a condition that results in dead or weakening body tissue. She sued the manufacturer Wyeth and won $6.8 million.
Another woman took the generic version of the defective drug, known as promethazine, and had to have her arm and forearm amputated because of complications from gangrene. Her case was dismissed last fall following the Supreme Court ruling.
The Supreme Court ruling comes as Americans are increasingly turning to generic medicines. As prices skyrocket and the economy struggles, many health insurance companies are requiring generics be filled before brand-name drugs. Doctors are required to show medical needs for the brand name over generic.
As a result, nearly 80 percent of prescriptions in the United States are filled generic and most states permit pharmacists to dispense a generic in place of a brand name.
What Can Consumers Do:
Support efforts to change the law. Public Citizen, a consumer advocacy group, has petitioned the FDA to give generic companies greater control over their labels. The move may allow generic drug users to sue. U.S. Rep. Henry A. Waxman, D-California, is also exploring ways to address the issue.
Talk to your doctor. Ask your doctor about the medicine being prescribed, the generic and potential side effects. If you are still concerned about potential injuries, ask your doctor to call your insurance company and request a brand-name.
Research any medication you use. Write down the name of the medicine you are prescribed, the medicine you receive at the pharmacy and research both drugs. Discuss any side effects with your physician.
Consider foregoing insurance. If you are really concerned and can afford the brand-name prescription, consider purchasing it. There are many discount drug programs which may help you reduce your costs. Check with any groups you are affiliated with, including AAA and AARP.
Contact your health insurance company. If the company has required you to use generic medications, ask if it has changed its policy and is now allowing use of brand-name medications.
Related:
Massachusetts Patients’ Bill of Rights and Preventing Medical Errors.
Product Recall: More than 10,000 Fuji Bicycles Recalled
 The U.S. Consumer Product Safety Commission announced the recall this week of more than 10,000 defective bicycles after reports the product’s frame was breaking.
The U.S. Consumer Product Safety Commission announced the recall this week of more than 10,000 defective bicycles after reports the product’s frame was breaking.
Fuji Saratoga Women’s Bicycles recalled about 10,500 bicycles sold nationwide from November 2007 through December 2011. The bicycles were recalled after the company became aware of 12 reports of bicycle frames breaking. There were two injuries reported, including a head laceration requiring 20 stitches.
The product’s defect is its frame was breaking in the center of the downtube during use, causing bicyclists to lose control and fall. Bicyclists are instructed to stop riding and seek a replacement bike frame.
About the recalled Fuji women’s cruisers bicycles:
- 2008 to 2010 models of Saratoga 1.0, Saratoga 2.0, Saratoga 3.0 and Saratoga 4.0. The model type will be printed on the bike.
- The bikes come in various colors.
- The bikes will have the words “Fuji” and “Saratoga” alone or “Saratoga” printed on the frame.
- Serial numbers beginning with ICFJ7, ICFJ8, ICFJ9, ICFJ10 and ICFJ11. The serial number is located on the bottom of the frame near the crank.
The defective products were imported by Advanced Sports, Inc. of Philadelphia and manufactured in China. They were sold at specialty bike shops.
Customers are instructed to obtain a free replacement frame. They can contact Advanced Sports Inc. toll-free at 888-286-6263 between 8 a.m. and 4:30 p.m. Monday through Friday or visit www.fujibikes.com. They can also return the bike to any authorized Fuji Bicycle dealer for the free part.
Click here for more information on this recall.
In a smaller recall, the Mountain Bicycle Handlebar Stem has been recalled in the U.S. and Canada. Some 213 units were recalled in the U.S. and 83 in Canada by the importer, Shimano American Corp. of Irvine, Calif. The defective bicycles were sold at REI stores nationwide from October 2009 to November 2010 for about $120.
The bicycles were recalled because the bolt holding the front plate of the stem to the stem body can be pulled out of the threads while the bike is being ridden, causing the rider to fall. There has been one report of a rider falling and sustaining torso and arm injuries. Click here for more information on this recall.
The Boston product liability lawyers at Breakstone, White & Gluck have over 80 years combined experience handling complex cases involving serious personal injuries, wrongful death and defective products. We have obtained clients compensation in cases involving defective motor vehicles, recalled medical devices and dangerous pharmaceuticals.
If you have been injured, it is important to learn your rights and how long you may have to file a claim. For a free legal consultation, contact us today at 800-379-1244 or 617-723-7676 or use our contact form.
Recalled Coffee Makers Burn Nearly 40 People
 More than 1.7 million coffee makers have been recalled after reports some machines have sprayed hot liquid, leaving 37 people with second-degree burns.
More than 1.7 million coffee makers have been recalled after reports some machines have sprayed hot liquid, leaving 37 people with second-degree burns.
The Tassimo Single-Cup Brewers were recalled Feb. 9 by BSH Home Appliances Corp. of Irvine, California. Some 835,000 machines were recalled in the United States and 900,000 were recalled in Canada. The California manufacturer recalled the defective product voluntarily along with the Consumer Product Safety Commission (CPSC) and Health Canada.
The brewers are defective because they can burst and spray hot liquid and coffee grounds or tea leaves onto consumers. There were a total of 140 reports of the brewers spraying hot liquid. Among the 37 second-degree burns was a 10-year-old Minnesota girl who suffered serious facial and neck burns which required her to be hospitalized.
The defective coffee makers carried the brand names of Bosch and Tassimo Professional Brewers. The Bosch brewers were sold in several colors to consumers between the dates of June 2008 and February 2012 for between $100 and $250. The Tassimo Professional was sold on in black, directly to hotels and food service providers. The brewers were manufactured in Slovenia and China.
Consumers are advised to stop using the recalled coffee makers immediately and contact the firm to order a free replacement T Disc holder part to fix the mechanism. This is the part of the machine that holds the single serve coffee cup.
Consumers can visit www.tassimodirect.com/safetyrecall for the full list of recalled models and to request a replacement part. They can also call the firm toll-free at 866-918-8763.
Click here to read the recall notice from the CPSC.
Read More
Multaq Heart Drug Warning from FDA
 After years of close monitoring and label changes for Multaq, the Food and Drug Administration (FDA) has issued a new warning that the heart drug places some patients at an increased risk for serious cardiac events including death.
After years of close monitoring and label changes for Multaq, the Food and Drug Administration (FDA) has issued a new warning that the heart drug places some patients at an increased risk for serious cardiac events including death.
Multaq is used to treat various conditions, including patients with permanent Atrial Fibrillation (permanent AF). This is the most serious form of AF, which is an abnormal heart rhythm. The two other types are paroxysmal AF and persistent AF.
Permanent AF is a chronic condition in which sinus rhythm cannot be sustained despite treatment.
The FDA issued its Dec. 19, 2011 safety notification based on its monitoring of a 10-year study. In the communication, the FDA stated Multaq doubles the rate of cardiovascular death, stroke and heart failure in patients with permanent AF. The FDA advised healthcare professionals to:
- Not prescribe Mutlaq to patients with AF who cannot or will not be converted into normal sinus rhythm (permanent AF).
- Monitor heart rhythm by electrocardiogram at least once every three months.
- If the patient is in permanent AF, Multaq should be stopped or the patient should be cardioverted.
- Multaq is indicated to reduce hospitalization for AF in patients in sinus rhythm with a history of non-permanent AF.
- Patients prescribed Multaq should receive antithrombatic therapy.
The study the FDA acted on was called called the Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS). The study was stopped early in July 2011. The FDA said it will release more information as it becomes available.
Multaq Background
Dronedarone is the generic name for Multaq. It was approved by the FDA in 2009 to treat several conditions, including permanent AF. Manufactured by Sanof-Aventis, the drug has been prescribed to 500,000 people around the world.
Since 2009, Multaq has been subject to several FDA actions, including in 2010 a revised warning noting cases of worsening heart failure in some patients. In February 2011, the FDA changed the warning label to state that Multaq should be discontinued if liver damage is suspected.
Related Blogs and Websites
- Actos: FDA Warning About Bladder Cancer
- DePuy Hip Recall: Insurers Seek to Collect Settlements
- Transvaginal Mesh: What to Know If You’ve Been Injured
Actos Warning From FDA
Hundreds of thousands of diabetes patients have been re-considering their medication plans after a recent warning from the Food and Drug Administration (FDA).
On June 15, 2011, the FDA issued a safety communication reporting that use of the oral diabetes medication Actos (pioglitazone) for more than one year may be associated with an increased risk of bladder cancer.
The FDA warned that patients with active bladder cancer should not be taking pioglitazone and those with a prior history should only use it after considering the potential medical benefits against the risk for cancer recurrence.
Actos is an oral drug used to treat type II diabetes, the most common form of the illness that plagues 25.8 million Americans. Actos is manufactured by Takeda Pharmaceutical Company and co-marketed by Eli Lilly and Company. It is one of the most widely used medications on the market. Over 2 million patients filled prescriptions for it in 2010.
The communication came as the FDA placed restrictions on Avandia, another popular medication, after studies showed it increased the risk of heart attacks. Both drugs belong to the thiazolidinedione class of drugs.
The warning about Actos came as the FDA made a periodic review of an ongoing 10-year Actos study into the risk for developing bladder cancer. The study reports there may be a 40 percent greater risk for developing bladder cancer among those who take Actos for more than a year.
The FDA’s warning came a new epidemiological study out of France suggested an increased risk of bladder cancer with pioglitazone use. As a result, France has suspended use of Actos and Germany has recommended new patients not be placed on the drug.
On August 4, 2011, the FDA reported it had approved new label and medication instructions for Actos. The FDA is continuing to monitor the results of Actos cancer studies.
Patients taking Actos should contact their doctors if they experience any sign of blood in the urine or a red color in the urine or other urinary pain, as these Actos complications may be signs of bladder cancer.
Click here to read the June 15, 2011 FDA Safety Communication on Actos.
Read More
DePuy Hip Implant Recall Leads Insurers to Seek Recovery
Medical insurance companies are alerting patients who have defective all-metal hip implants that they will seek to recover expenses from any settlement money which patients receive. Medicare is also expected to try and recover its costs.
Thousands of patients have been affected by recent recalls of all-metal hip implants and are expected to seek compensation from manufacturers. Although insurance companies, by statute or contract, often have the right to recover some of their costs, it is unusual for companies to be so assertive for medical device failures. The aggressive actions being taken by the insurers reflect their awareness of just how expensive these defective products are becoming.
In August 2010, DePuy recalled the ASR XL Acetabular System. The hip implant is defective because it causes friction between the metallic ball and socket components, which are implanted to replace the femur and acetabulum.
The recalled DePuy hip implants can wear down and produce a substantial amount of metallic particles in patients’ bloodstreams. Patients reported signs of metallosis, including pain, swelling, problems walking and rashes. Other serious problems may include bone fractures, joint inflammation, hip dislocation and damage to the tissue, hip implant loosening, nerves and muscles near the implant.
DePuy recalled the defective hip implants after the Food and Drug Administration (FDA) received about 400 complaints in two years from patients. That number has grown significantly following the recall. In just the first six months of 2011, the FDA received more than 5,000 reports about problems with all-metal hip implants, according to an analysis by The New York Times. DePuy hip implants accounted for 75 percent of the complaints.
It is still unclear how many patients have been affected by defective metal hip implants. The New York Times reports one estimate that 500,000 patients have received an all-metal replacement hip.
The newspaper reports another estimate that 250,000 hip replacements are performed in the United States each year. Until recently all-metal hip implants accounted for nearly one-third of these procedures.
As of October 2011, 3,500 patients filed lawsuits against DePuy in connection with last year’s recall. DePuy also faces over 560 lawsuits in connection with the Pinnacle, another defective all-metal hip implant.
More lawsuits are expected against DePuy and other manufacturers as patients start experiencing pain and requiring corrective surgery. This procedure can be painful, require substantial post-operation bed rest and treatment and cost hundreds of thousands of dollars.
Click here to read more about all-metal hip implants in the New York Times.
Read More
Infant’s Death Prompts Walmart To Remove Baby Formula
 Walmart pulled Enfamil baby formula from more than 3,000 stores this week after a newborn’s death in Missouri.
Walmart pulled Enfamil baby formula from more than 3,000 stores this week after a newborn’s death in Missouri.
Walmart voluntarily removed the cans of Enfamil Newborn from its store shelves on Monday night. The baby formula is under investigation by health officials after the 10-day-old baby boy’s death Sunday from Cronobacter, a bacteria linked to newborn illness and milk-based powder baby formula. The formula had been used by the baby’s family. A second infant was infected by the bacteria, but recovered.
The 12.5-ounce cans of Enfamil Newborn are marked with the lot number ZPIK7G. This includes at the chain’s 35 stores, 12 super centers and 2 Sam’s Club locations in Massachusetts.
The formula has been sent for testing to the U.S. Centers for Disease Control and Prevention (CDC) and the Food and Drug Administration (FDA). In the meantime, the Missouri Department of Heath and Senior Services is urging consumers who purchased the formula to return it to the store or discard it.
The manufacturer Mead Johnson said that the batch of infant formula used by the child’s family tested negative for Cronobacter when it was produced and packaged.
Mead Johnson and Walmart representatives say the companies are working with authorities. Neither of the companies has implemented a formal product recall. The government has not requested one.
Click here to read a recent news article about this product liability case.
Read More
Medical Device Recall for CooperVision Contact Lenses
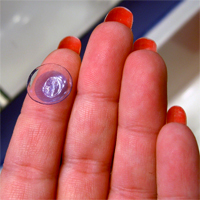 Consumers are advised to stop using a second type of CooperVision’s Avaira contact lenses due to a defect that may result in blurry vision and other eye injuries.
Consumers are advised to stop using a second type of CooperVision’s Avaira contact lenses due to a defect that may result in blurry vision and other eye injuries.
On Nov. 16, an unknown quantity of CooperVision Avaira (enfilcon A) Sphere contact lenses were recalled due to the unintended presence of a silicone oil residue.
CooperVision, a Fairport, NY-company, recalled the lenses in cooperation with the Food and Drug Administration (FDA), which states that the silicone oil residue can result in a wide range of symptoms, including eye discomfort, hazy and blurry vision and eye injuries requiring medical treatment. The FDA regulates contact lenses as a medical device.
The recalled lenses were manufactured from Feb. 1, 2011 to Aug. 24, 2011 and distributed from March 2, 2011 to Nov. 15, 2011.
The contact lenses were used to correct myopia and hyperopia and non-aphakic individuals with non-diseased eyes. The lenses may be worn by individuals who have astigmatism of 2.00 diopters or less that do not interfere with visual acuity.
Lens wearers have been instructed to stop using the lenses immediately and contact their eye care professional. They can visit the CooperVision recall website and enter the defective medical device package numbers to determine if their lenses have been recalled.
The medical device recall expanded another in August 2011 for limited lots of Avaira Toric contact lenses. CooperVision said the product defect on that line has been corrected through its quality system process.
Contact lens injuries are prevalent, due to product defect or improper use. They are most common among children and adolescents. In a study published in the July 2010 Pediatrics, FDA researchers reported 23 percent of total medical device injuries in 2004 and 2005 in the U.S. involved children and contact lenses. Children ages 11 and over were the most affected.
Common personal injuries include corneal contusions and abrasions, hemorrhage and conjunctivitis.
Click to read the FDA notice about the CooperVision recall.
Read More
Food Recall for Ocean Spray Dried Cranberries
 As the Christmas season begins, one Massachusetts company remains bogged down in Thanksgiving, after issuing a recall for its popular dried cranberry snacks.
As the Christmas season begins, one Massachusetts company remains bogged down in Thanksgiving, after issuing a recall for its popular dried cranberry snacks.
The day after Thanksgiving, Ocean Spray of Middleboro issued a recall for four package sizes of its Original Flavor Craisins Dried Cranberries product. The company voluntarily recalled the snack because of the possibility packages contained small hair-like metal fragments. The metal fragments resulted from an equipment malfunction on a production line, a company official told Vitals, an MSNBC.com blog.
Ocean Spray said it has received no consumer complaints or injury reports. Dried cranberry snacks are a growing product line for Ocean Spray. Last summer, a company official told the New England Business Bulletin that it was planning to expand its Middleboro facility and produce a third dried cranberry product.
The following Ocean Spray Original Flavor Craisins Dried Cranberries are part of this product recall:
- 5 oz Craisins® UPC: 00293-000
- 10 oz Craisins® UPC: 29456-000 and 29464-000
- 48 oz Craisins® UPC: 00678-318
- 10 lb bulk ingredient & foodservice UPC: 03477-000
Click here to see the full list with production dates for the recalled products.
Consumers who have purchased the recalled products are advised to discard them and preserve the UPC label and best by date. They should contact the Ocean Spray Consumer Hotline at 1-800-662-3263 for a coupon replacement.
Read More