

Posts Tagged ‘“medical devices”’
Medical Device Recall for CooperVision Contact Lenses
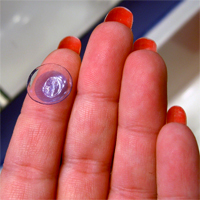 Consumers are advised to stop using a second type of CooperVision’s Avaira contact lenses due to a defect that may result in blurry vision and other eye injuries.
Consumers are advised to stop using a second type of CooperVision’s Avaira contact lenses due to a defect that may result in blurry vision and other eye injuries.
On Nov. 16, an unknown quantity of CooperVision Avaira (enfilcon A) Sphere contact lenses were recalled due to the unintended presence of a silicone oil residue.
CooperVision, a Fairport, NY-company, recalled the lenses in cooperation with the Food and Drug Administration (FDA), which states that the silicone oil residue can result in a wide range of symptoms, including eye discomfort, hazy and blurry vision and eye injuries requiring medical treatment. The FDA regulates contact lenses as a medical device.
The recalled lenses were manufactured from Feb. 1, 2011 to Aug. 24, 2011 and distributed from March 2, 2011 to Nov. 15, 2011.
The contact lenses were used to correct myopia and hyperopia and non-aphakic individuals with non-diseased eyes. The lenses may be worn by individuals who have astigmatism of 2.00 diopters or less that do not interfere with visual acuity.
Lens wearers have been instructed to stop using the lenses immediately and contact their eye care professional. They can visit the CooperVision recall website and enter the defective medical device package numbers to determine if their lenses have been recalled.
The medical device recall expanded another in August 2011 for limited lots of Avaira Toric contact lenses. CooperVision said the product defect on that line has been corrected through its quality system process.
Contact lens injuries are prevalent, due to product defect or improper use. They are most common among children and adolescents. In a study published in the July 2010 Pediatrics, FDA researchers reported 23 percent of total medical device injuries in 2004 and 2005 in the U.S. involved children and contact lenses. Children ages 11 and over were the most affected.
Common personal injuries include corneal contusions and abrasions, hemorrhage and conjunctivitis.
Click to read the FDA notice about the CooperVision recall.
Read More
Defective Product Leads to Proposed Plea Deal in the Largest Criminal Penalty Ever Assessed Against a Medical Device Company
Guidant LLC, a division of Massachusetts’ company Boston Scientific, has plead guilty to two misdemeanor counts alleging the medical device maker failed to disclose product changes involving over 20,000 implantable heart monitor devices.
The medical device manufacturer plead guilty Monday, April 5 and will learn over the next few weeks whether U.S. District Judge Donovan Frank will accept a proposed $296 million plea deal – the largest criminal assessment ever proposed against a medical device company.
The Department of Justice accuses Guidant of changing the design of its implantable cardioverter defribrillators, or ICDs, and failing to notify the Food and Drug Administration (FDA) of subsequent problems that lead to a Class 1 medical recall – the most serious category which indicate a defective product has the potential to cause serious personal injury or wrongful death.
Guidant’s implantable cardioverter defribrillators, Ventak Prizm 2 DR and Contak Renewal 1 and 2, were designed to monitor patients for abnormal heart rhythms and deliver electric shocks to keep the heart beating properly. But Department of Justice officials say Guidant discovered as early as 2002 that Ventak had the potential to suffer an electric arc, which could short-circuit the device. Although problems continued with the defribrillator, Guidant didn’t issue a warning until 2005. In at least seven cases, the devices failed to issue a lifesaving shock and the patient died.
In 2005, Guidant sent a product update to doctors, advising that a yellow warning screen indicated a potentially serious problem. However, the FDA says the company should have sent a product correction, rather than a product update, since the change reduced the risk of serious injury, and should have notified the FDA of the change within 10 days. Guidant ultimately recalled its three devices in 2005.
Attorneys for the affected patients are now urging the court to reject the plea deal because it will not provide restitution payments to victims. The government prosecutor has argued that the victims have other remedies for compensation and that the applicable law does not require restitution. The prosecutor points to the fact that most of the victims have settled civil suits with the company and the company has paid out over $650 million in settlement and warranty payments. Additionally, $42 million of the plea amount is forfeited funds and victims can petition the Justice Department for their share.
For more information on the plea deal, see this Boston Globe article and this Star Tribune article.
CALAXO Screw Recall–Defective Surgical Screws Recalled by Manufacturer, Smith & Nephew
The CALAXO surgical screw has been recalled because it has been identified as a dangerous medical device. The device, which has been used in surgical repairs of the anterior cruciate ligament (ACL) during knee surgery, has been determined to be a defective product because of the unacceptably high rate of post-surgery complications. The defective product has been approved for use in the United States since 2006,has been recalled by the United States distributor, Smith & Nephew, Inc., Endoscopy Division of Andover, MA. The US recall was issued urgently on August 21, 2007. The screw was also ordered recalled in the United Kingdom.
If you had surgery which involved the CALAXO surgical screw, you should
contact the CALAXO screw recall lawyers at Breakstone, White & Gluck to determine your rights and options on this defective product. Time is of the essence in product liability cases.
More Details on the Defective Product: The CALAXO Bioabsorbable Interference Screw is used to secure the anterior cruciate ligament graft during surgical repair of the knee ligament. The screw is designed to be absorbed by the body within a year following the procedure and also to promote bone
growth. In some patients, the CALAXO screw caused tissue swelling in the tibia, where the screw has been placed. Symptoms may include swelling, redness, fever, and severe pain.